МЕХАНИЗМЫ РАЗВИТИЯ АТЕРОСКЛЕРОЗА ПРИ ОЖИРЕНИИ
В настоящее время атеросклероз рассматривается как системное заболевание, характеризующееся формированием одиночных и множественных атероматозных бляшек (очагов липидных, главным образом – холестериновых, отложений) на внутренней поверхности крупных артерий, приводящих к нарушению их проходимости. Изменения структуры стенок сосудов в результате отложения холестерина стимулирует процессы локального воспаления, сопровождающиеся постепенным развитием соединительной ткани (склерозом) и отложением солей кальция (кальцинозом), существенно усугубляющими сужение просвета артерий и нарушение тока крови по ним.
Термин “атеросклероз” (“athtre” – пшеничная каша, “sclerosis” - твердый) предложен в 1904 году F. Marchand. Этиология (причины) и патогенез (процесс развития) заболевания исключительно сложны и не до конца изучены. Отмеченное обстоятельство объясняет повышенный интерес к проблеме не только среди специалистов практической и теоретической медицины, но и исследователей, работающих в различных областях фундаментальной науки: химии, физики, биологии и др. Изучение тонких механизмов развития атеросклероза неразрывно связано с пониманием таких глубоких и принципиальных вопросов функционирования живых многоклеточных организмов, как передача наследственной информации, старение, развитие системных заболеваний и злокачественных новообразований и многих других нерешенных задач человечества.
Значительное количество “белых пятен” в современных представлениях о проблеме является причиной существования конкурирующих взглядов на механизмы возникновения и прогрессирования атеросклероза, имеющих свои сильные и слабые стороны, однако, в общих чертах, укладывающихся в две основные концепции.
Нарушения процессов регуляции метаболизма липидов (дислипидемий), и в первую очередь - обмена холестерина, составляют основу “инфильтративно-гиперпластической” или “холестериновой” теории (Н. Н. Аничкова и С.С. Халатова). В течении века существования данной концепции первоначальные теоретические положения получили убедительные подтверждения ее научной обоснованности и стали базой для дальнейшего изучения механизмов развития заболевания с позиций современных знаний.
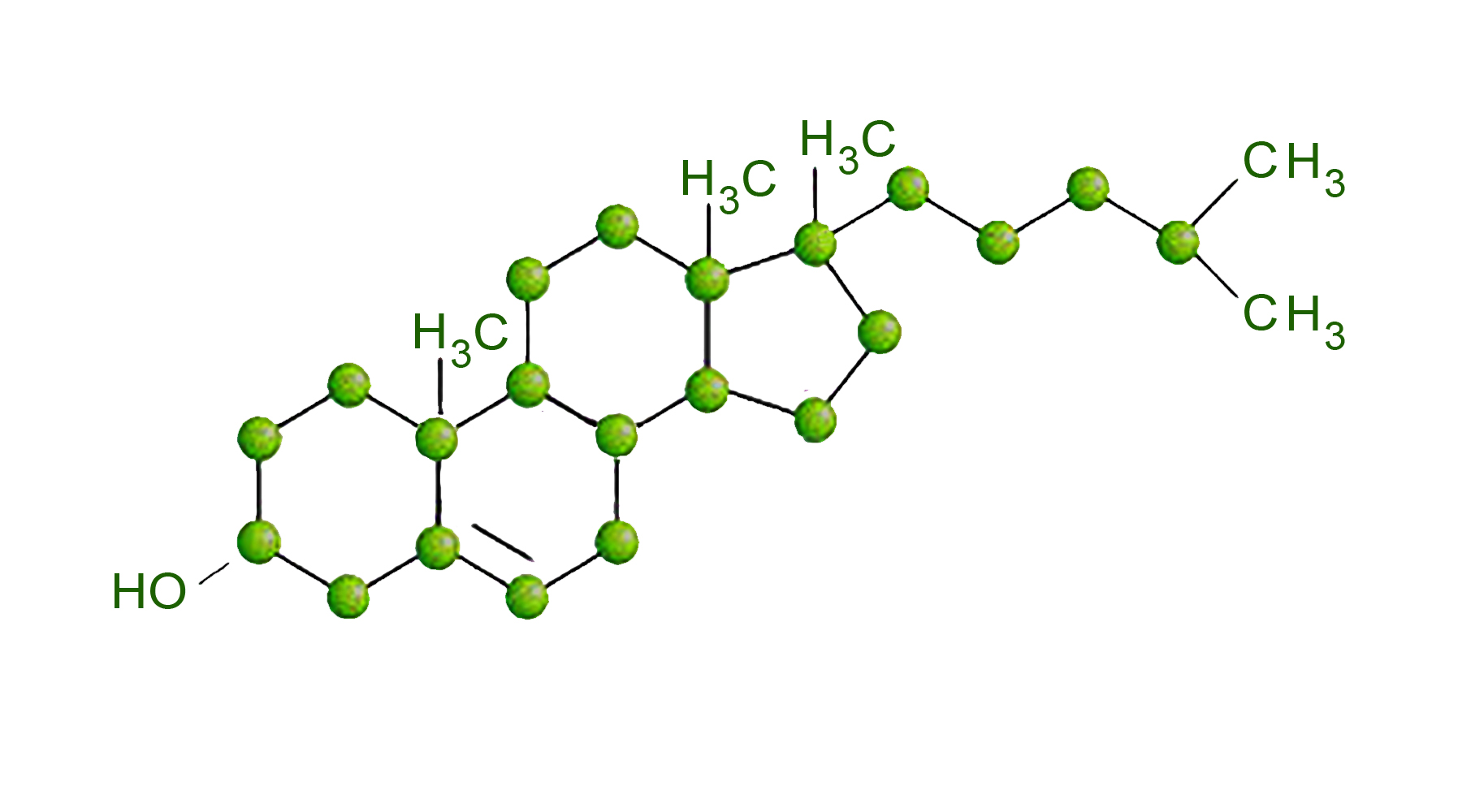
Строение молекулы холестерина
К настоящему времени получено множество доказательств, что липиды, в том числе холестерин и его эфиры, располагающиеся внутри и вокруг клеток интимы (внутренней оболочки) и медии (средней части стенки) артерий и участвующие в образовании атеросклеротических бляшек, происходят из липопротеинов плазмы крови.
Липидный спектр плазмы человека составляют триглицериды (эфиры жирных кислот и глицерина), фосфолипиды (сложные производные жирных кислот и фосфорной кислоты), эфиры холестерина (соединения жирных кислот и холестерина), а также свободные (неэстерифицированные) жирные кислоты.
Эфиры холестерина в качестве обязательного липидного компонента входят в состав липопротеинов (комплексных соединений с белками - апопротеинами), представляющих универсальный механизм транспорта и передачи тканям различных производных жирных кислот. В зависимости от состава и размеров различают липопротеины очень низкой плотности (ЛПОНП), низкой плотности (ЛПНП), высокой плотности (ЛПВП), липопротеиды средней плотности (ЛПСП) и хиломикроны. Каждая группа липопротеинов очень неоднородна по размерам частиц, а также содержанию апопротеинов и липидов. Основная часть холестерина переносится липопротеинами низкой плотности, существенно меньшая – ЛПОНП и липопротеинами высокой плотности. В отличие от холестерина, эндогенные триглицериды транспортируются преимущественно в составе ЛПОНП.
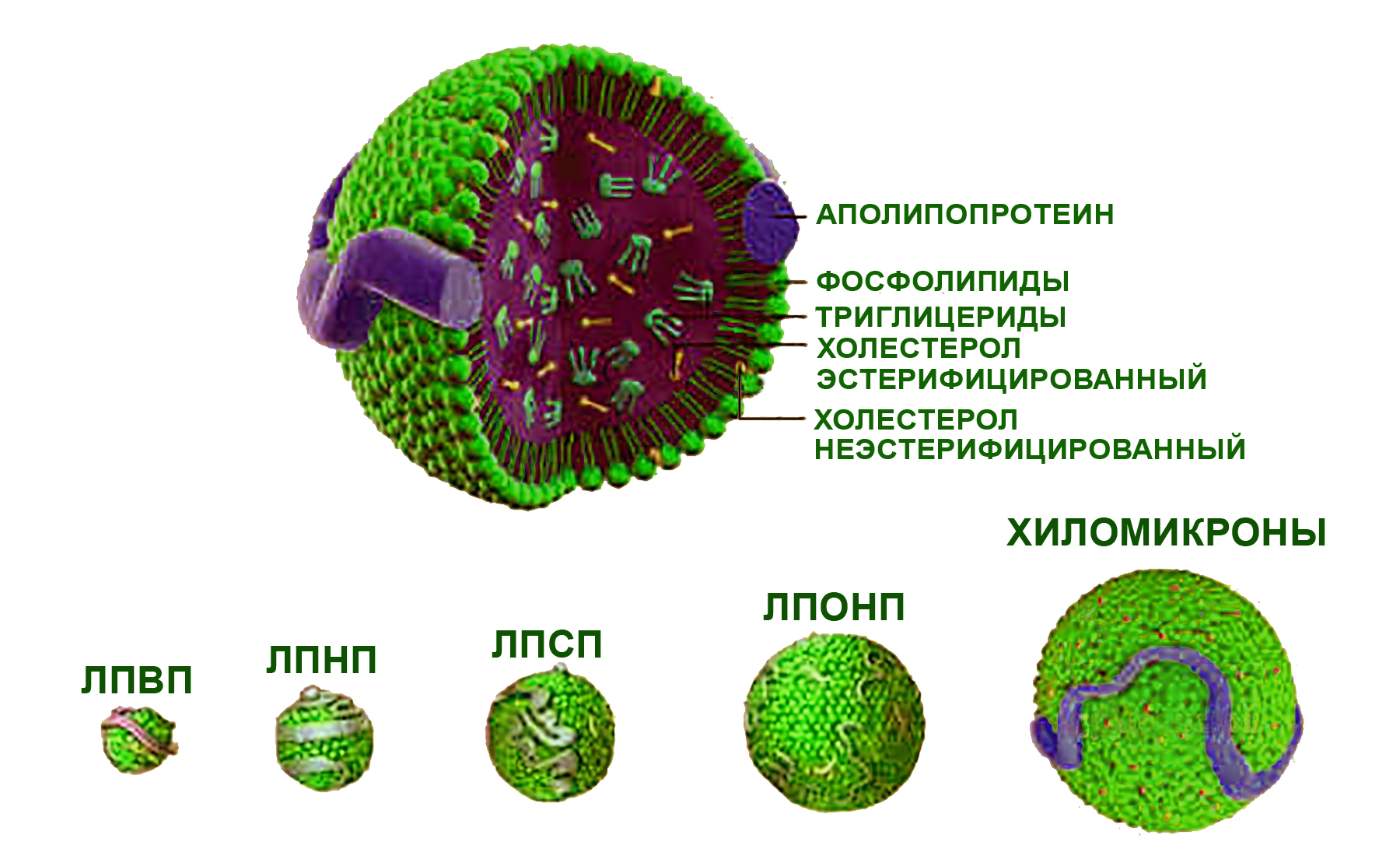
Строение и соотношение размеров липопротеинов
Функцией апопротеинов, входящих в состав липопротеинов, являются повышение растворимости (в плазме крови) эфиров холестерина и триглицеридов, регуляция и кооперация взаимодействия липидов с ферментами (в процессе биохимических реакций) и связь с рецепторами при транспорте соединений жирных кислот в клетки тканей.
С позиций современных знаний внутриклеточный и тканевой метаболизм липопротеинов разных классов упрощенно можно представить следующим образом. Хиломикроны, образующиеся в тонкой кишке из липидов пищи, поступают через лимфу в плазму крови, собирающуюся через систему воротной вены печенью. Под действием ферментов плазмы крови (липопротеинлипазы) хиломикроны распадаются на более мелкие комплексы (ремнанты), которые и захватываются гепатоцитами (клетками печени). Синтезированные в печени триглицериды в составе ЛПОНП поступают в плазму, где они, как и хиломикроны, претерпевают частичную деградацию до ремнантных липопротеинов низкой и особо низкой плотности. Циркулирующие в плазме ЛППП (липопротеины промежуточной плотности) и ЛПОНП утилизируются клетками различных тканей, а при их избытке – подвергаются катаболизму (разложению) в печени до триглицеридов. Липопротеины высокой плотности образуются из холестерина, синтезированного клетками печени и других тканей, или поступившего с пищей, и компонентов (фосфолипидов) хиломикронов и ЛПНП.
Столь подробное изложение сложных процессов синтеза, транспорта, резервирования и утилизации производных жирных кислот в организме позволяет представить многогранность возможных причин нарушения механизмов их регуляции, играющих роль в развитии дислипопротеинемии (изменении концентрации различных липопротеинов в плазме крови).
Роль генетических факторов в возникновении атеросклероза в настоящее время не вызывает сомнений и доказана существованием как минимум трех описанных патологических состояний (дислипопротеинемий): семейной гиперхолестеринемии, семейной комбинированной гиперлипидемии и семейной гипертриглицеридемии.
Семейная гиперхолестеринемия является следствием мутации гена, контролирующего синтез рецепторов к ЛПНП, приводящих в итоге к повышенному синтезу холестерина и ЛПОНП. Данный вариант наследственной дислипидемии приблизительно в трети случаев характеризуется злокачественным течением атеросклероза и практически не поддается консервативной терапии.
Семейная комбинированная гиперлипидемия вызывается доминантным (подавляющим проявление другого, отвечающего за тот же признак) геном. У людей с быстрым развитием атеросклероза на долю данной мутации приходится 15% случаев.
Третьим из наиболее распространенных наследственных заболеваний, приводящих к быстрому развитию атеросклероза, является семейная гипертриглицеридемия. Мутация доминантного гена проявляется значительным повышением концентрации в плазме крови ЛПОНП и снижением уровня ЛПВП. Данный тип генетических изменений отмечается у 5% больных.
Наследственные варианты развития атеросклероза отмечаются менее чем у 10% пациентов с данным заболеванием. Большая же часть гиперлипопротеинемий относится к категории приобретенных. Причиной их возникновения является совокупность внешних факторов (характера питания, приема некоторых медикаментозных препаратов, сопутствующих заболеваний и т.д.) и его генетических особенностей (к сожалению, многочисленных и не до конца понятных). Сложный комплекс взаимодействий между указанными экзогенными (внешними) и эндогенными (внутренними) факторами может приводить к нарушению обмена липидов, и в итоге – к развитию атеросклероза.
Развитие атеросклероза зависит не только от абсолютного уровня тех или иных классов липопротеинов, но и от соотношения липопротеинов с атерогенной (вызывающей атеросклероз) и антиантиатерогенной (препятствующей возникновению заболевания) направленностью. Ярко выраженным атерогенным эффектом обладают ЛПНП и ЛПОНП, в то время как липопротеины высокой плотности препятствуют развитию заболевания.
Таковы общие представления о роли нарушений липидного обмена в возникновении и прогрессировании атеросклероза с позиций “холестериновой” теории.
Однако неверным было бы рассмотрение патогенеза данного состояния исключительно с точки зрения дисбаланса между различными липопротеинами в плазме крови, поскольку характерные для атеросклероза изменения происходят именно в стенках крупных артерий.
Начальными признаками развития заболевания являются набухание и разрыхление внутреннего слоя (эндотелия) кровеносных сосудов. Изменение свойств эндотелия (которое может вызываться повышением артериального давления, воздействием вирусов, бактерий, эндо- или эзогенных токсинов и т.д.) приводит к агрегации и адгезии (концентрации и прилипанию) тромбоцитов, являющимися начальной фазой физиологической реакции коагуляции (свертывания) крови и формирования микротромбов при нарушении целостности сосудов. Параллельно с процессом агрегации тромбоцитов происходит синтез фибрина, нити которого являются “каркасом” для клеточных элементов тромба. Продуцируемые тромбоцитами биологически активные вещества (цитокины) инициируют процесс воспаления, усиливают образование первоначального тромба и вызывают адгезию других клеточных (лимфоцитов, моноцитов, макрофагов и др.) и молекулярных компонентов плазмы крови, включая атерогенные липопротеины. Способностью к эндоцитарному (внутриклеточному) захвату частиц ЛПНП и ЛПОНП под действием медиаторов воспаления могут обладать клетки эндотелия и других слоев стенки артерий, а также - присутствующие в зоне тромба макрофаги (специфические клетки, уничтожающие в организме чужеродные агенты).
С течением времени тромбоциты, выполнившие свою физиологическую функцию, подвергаются распаду и утилизации (макрофагами), а в зоне первоначального повреждения сосуда формируется атеросклеротическая бляшка, состоящая из нитей фибрина (соединительной ткани) и отложений холестерина. Воспалительные изменения приобретают характер хронических (постоянных) вследствие повреждающего действия турбулентного (вихревого) потока крови в области нарушения структуры внутренней поверхности артериальной стенки и изменения ее естественного строения. Воспалительный процесс стимулирует гибель и заместительную пролиферацию миоцитов (клеток мышечных клеток мышечного слоя), развитие соединительной ткани, отложение солей кальция (кальциноз), в итоге приводящих к еще более выраженному локальному утолщению и разрыхлению стенки с сужением просвета сосуда.
Описанные изменения проходят ряд последовательных стадий.
- Образование липидного пятна (или полоски), представляющего собой участки бледно-желтого цвета, содержащие липиды, не возвышающиеся над поверхностью интимы артерии.
- Формирование фиброзной бляшки – овального или округлого образования, содержащего липиды, возвышающегося над поверхностью интимы и нередко сливающегося в сплошные бугристые поля.
- Кальциноз – отложение в фиброзной бляшке солей кальция.
- Деструктивные изменения фиброзной бляшки: изъязвление, кровоизлияние, наложение тромботических масс.
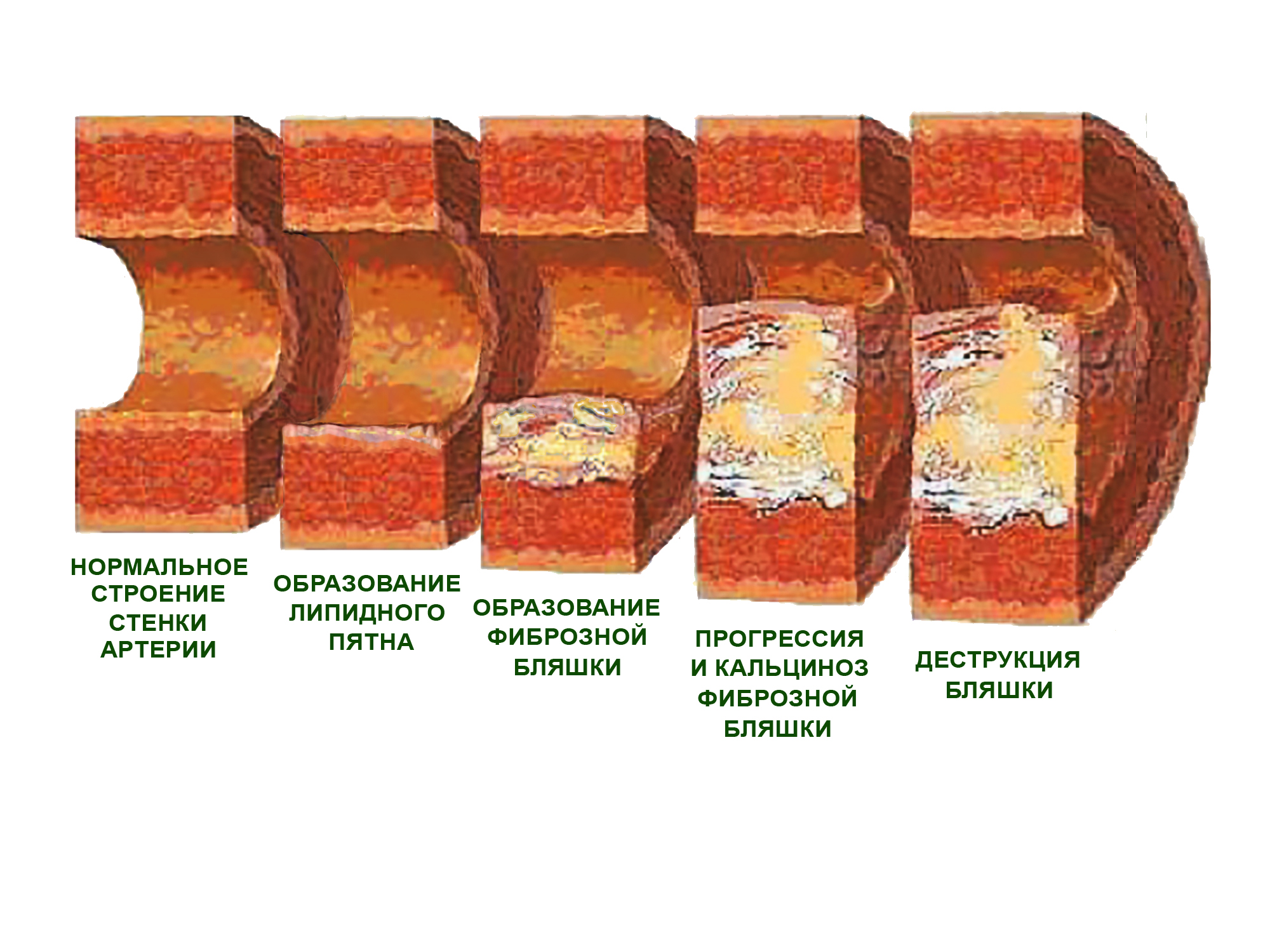
Стадии развития атеросклеротической бляшки
Таковы основные положения “эндотелиальной” концепции развития атеросклероза. Однако так же, как “холестериновая” теория, данная гипотеза имеет ряд слабых мест и не позволяет ответить на ключевой вопрос: почему, при прочих равных условиях, в одних случаях заболевание возникает, а в других – нет?
Современные представления о проблеме позволяют считать, что развитие атеросклероза происходит с участием изложенных в свете обоих теорий процессов. Вероятно, в развитии заболевания принимают участие и многие другие, пока не изученные, механизмы.
Однако даже существующие на сегодняшний день взгляды дают возможность очертить круг внешних и внутренних факторов, несущих риск возникновения атеросклероза, и сформировать комплексную систему профилактики и лечения данного системного патологического процесса и его осложнений.